Information on Sickle Cell Disease
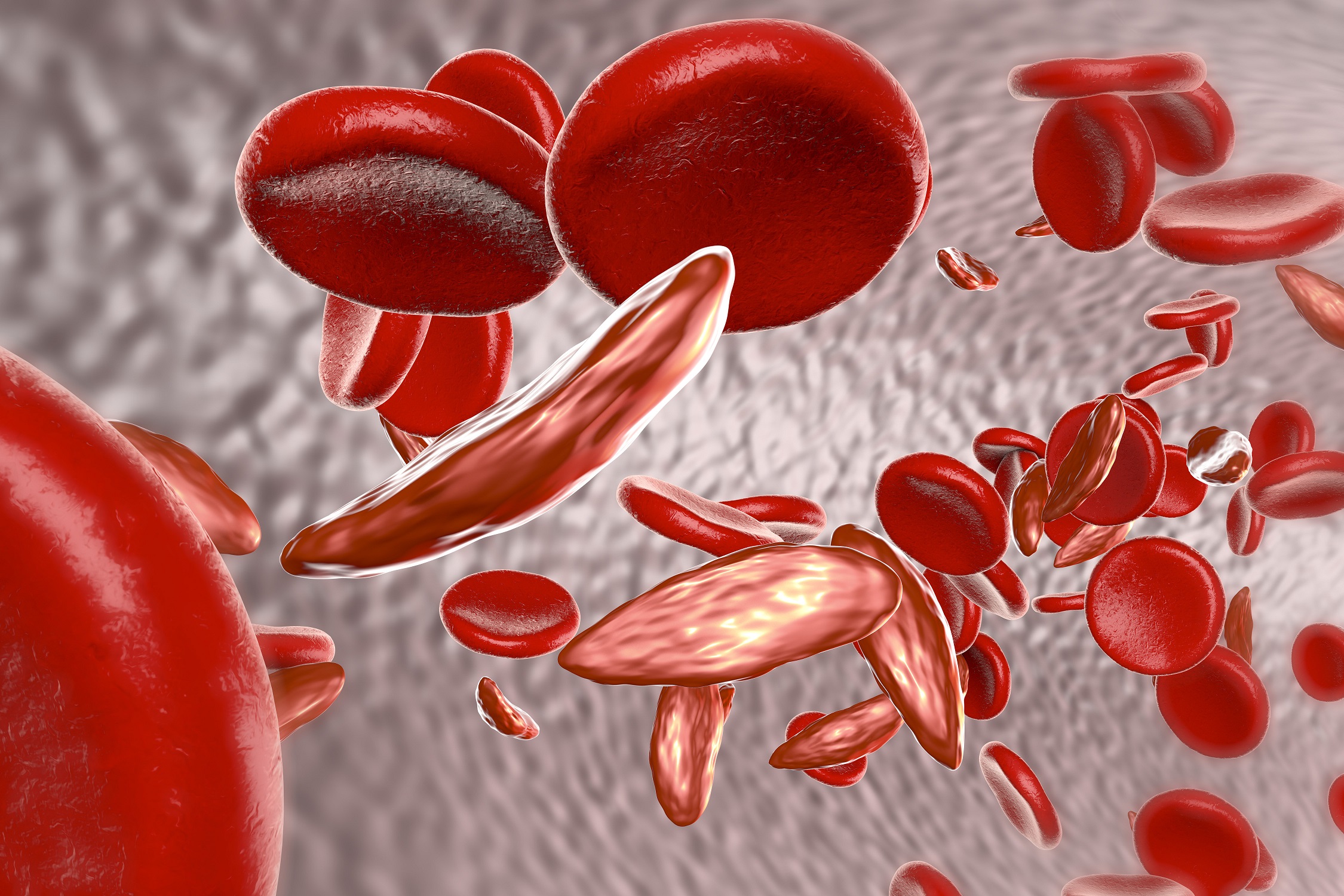
Sickle cell disease is a disease of the red blood cells (erythrocytes). Because of a change in the genetic material of the affected person, the erythrocytes lose their round and smooth shape and become hard and immobile cells, which we call sickle cells.
The role of the red blood cells
The main function of red blood cells (erythrocytes) is to transport and deliver oxygen to the respective target tissue or organ in the body. For this purpose, the oxygen in the lungs is bound to the red blood pigment (haemoglobin).
Healthy erythrocytes contain normal haemoglobin (HbA). This consists of two different protein chains whose spatial arrangement determines the structure and function of the haemoglobin. These subunits are called alpha and beta chains.
In sickle cell disease, the beta chain in haemoglobin is changed. In this context we speak of sickle cell haemoglobin (HbS). This causes the erythrocytes to lose their round shape and deform into sickle-shaped cells.
Disease history: Why are sickle cells a problem?
The change in the shape of the erythrocytes has an influence on their function. In order to be able to supply every region of the body and all organs with oxygen, the erythrocytes must be deformable and smooth. This enables them to pass through even the smallest blood vessels.
The sickle cells are less mobile than healthy erythrocytes. They get stuck in small blood vessels and interrupt the oxygen supply to the relevant tissue. The resulting reduced blood flow can cause damage to which the body reacts by producing substances that cause pain.
In addition, sickle cells have a shortened lifespan compared to healthy erythrocytes. Whereas healthy erythrocytes survive for 120 days on average, sickle cells are broken down after 10 to 20 days. In sickle cell patients, as a result, this degradation outweighs the production of new cells. This results in chronic anaemia, which leads to a lack of oxygen.
Frequency: How many people suffer from sickle cell disease?
Sickle cell disease is one of the most common hereditary diseases worldwide. It first appeared in Central Africa and on the Arabian Peninsula. It is assumed that sickle cell disease has been able to spread in these countries because the carriers of the sickle cell gene have a selective advantage over a severe and deadly form of malaria (Malaria tropica). They are better protected against this form of malaria and survive more often than people without a defective genetic make-up. This protection, however, only concerns people with carrier status and sickle cell patients who do not have the disease.
From these areas, sickle cell disease first spread to the Mediterranean region and North America through trade links and migration. In the meantime, the disease is also present in Northern Europe. In Germany there are currently about 3000-5000 affected people.
Causes: Why does sickle cell disease develop?
Sickle cell disease is a hereditary disease that is not contagious. Those affected have inherited a genetic disposition for the altered blood pigment haemoglobin from their parents.
A distinction is made between carriers and sufferers. If a child inherits the altered genetic make-up for haemoglobin from both parents, this leads to the development of sickle cell disease. Carriers of the genetic disposition however, have inherited the disposition (for example for HbS) from only one parent. They therefore carry one healthy and one diseased gene.
Persons with carrier status do not develop symptoms of sickle cell disease because they produce healthy haemoglobin (HbA) and sickle cell haemoglobin (for example HbS). However, they can transmit the sickle cell gene to their own children.
Patients and carriers of sickle cell disease are advised to seek genetic counselling if they wish to have children. During such a consultation, the risks and possibilities for the birth of a healthy child can be discussed.
Symptoms: What symptoms do affected persons have?
Most complaints arise as a result of the immobility and short life of the sickle cells. However, the symptoms and their severity can vary from person to person.
Typical for sickle cell disease are phases of severe pain, which can affect the skeletal system or organs. There are several factors that can trigger a so-called pain crisis. These include changes in the weather and sudden changes in body temperature as well as a lack of fluids, physical exhaustion, stress and infections.
Clumps in the blood vessels can lead to damage to the cardiovascular system (acute thorax syndrome), the central nervous system (strokes) and the spleen (splenic death).
The function of the spleen, which filters pathogens from the blood, can be impaired by blocked blood vessels in the first years of life. For this reason, sickle cell patients have an increased risk of serious bacterial infections.
Children with sickle cell disease are also at risk of splenic sequestration. This is a life-threatening condition in which large quantities of blood are stored in the spleen and are no longer available to the rest of the body.
Because of the large amount of blood, the spleen swells and can be palpated. Parents of children with sickle cell disease are recommended to learn how to palpate (feel) the spleen and to use this technique regularly.
A complete and more comprehensive description of possible symptoms of sickle cell patients can be found on the information portal kinderblutkrankheiten.de.
Diagnostics: Which examination can be used to determine sickle cell disease?
In the USA and some European countries, new-born babies are screened for sickle cell disease shortly after birth. With this routine examination, the disease can be detected early.
Routine screening is not yet performed in Germany. The diagnosis can only be made after the appearance of symptoms.
Therapy: How can sickle cell disease be treated?
Sickle cell disease is a serious disease that accompanies the affected person throughout his or her life. The quality of medical care has a decisive influence on the quality of life of those people affected. Therefore, we recommend that patients are cared for by the experts.
For further information on therapy options, please contact the doctor who is treating you.
More information on sickle cell disease can be found on the pages of the Sickle Cell Disease and Thalassaemia Interest Group (www.ist-ev.org) and on the kinderblutkrankheiten.de (www.http://www.kinderblutkrankheit.de) information portal.


